Tabla de contenidos
ToggleEl síndrome de Usher es una condición poco frecuente que afecta a dos sentidos esenciales: la audición y la visión. Aunque muchas veces pasa desapercibida en sus etapas iniciales, con el tiempo puede provocar alteraciones visuales que impactan de manera significativa en la vida diaria.
Esta enfermedad de origen genético no solo plantea desafíos auditivos, sino también oftalmológicos, ya que puede alterar progresivamente la capacidad visual y requerir un seguimiento especializado. Comprenderla es el primer paso para detectar a tiempo sus manifestaciones oculares y proteger la salud de los ojos.
En este artículo conocerás qué es el síndrome de Usher, cuáles son sus principales causas, cómo se diagnostica y qué opciones existen para mantener una buena visión a pesar de su avance. Sigue leyendo y descubre por qué la detección temprana puede marcar una gran diferencia.
▶ ¿Necesitas una evaluación ocular? Reserva una cita en Opeluce.
¿Qué es el síndrome de Usher?
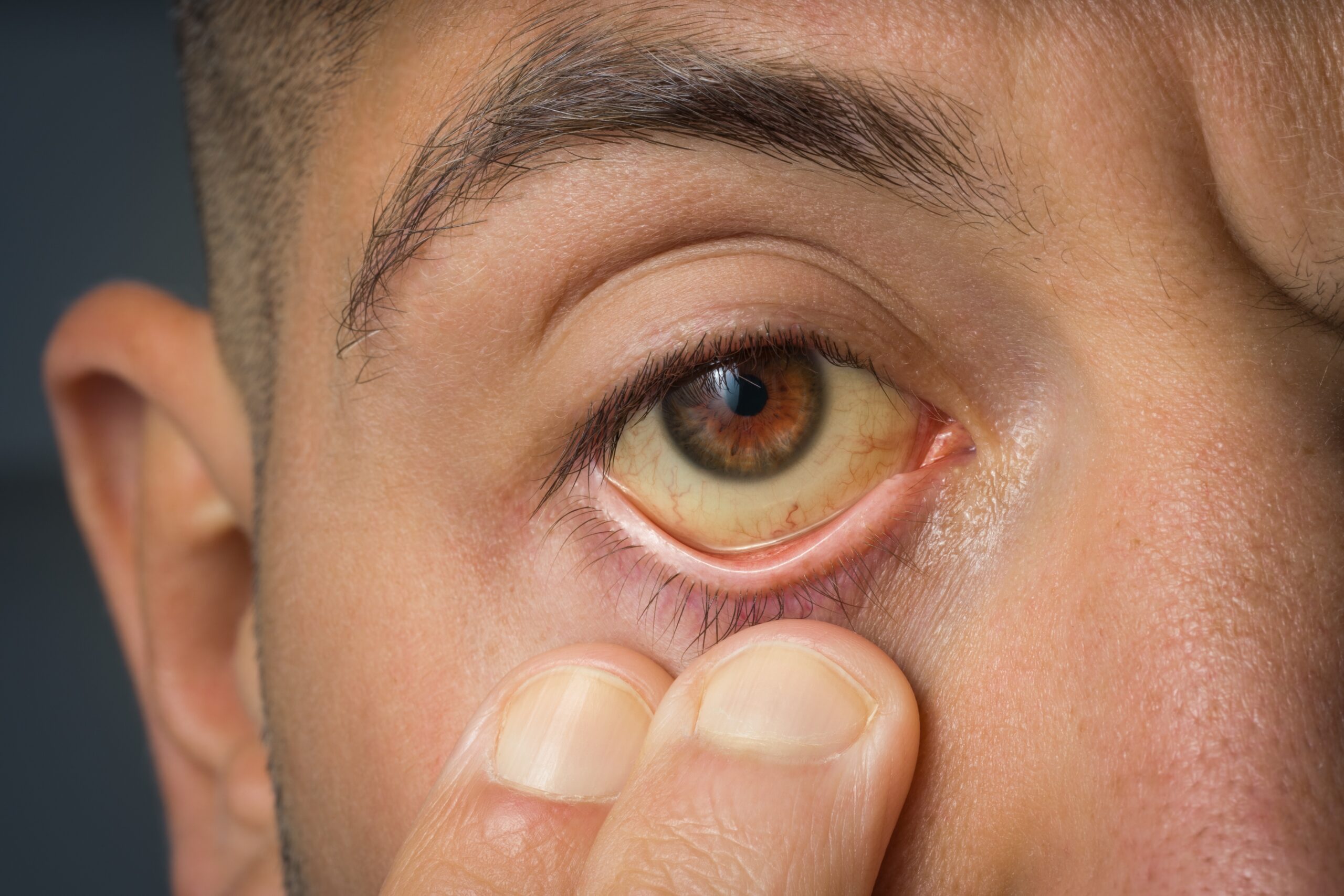
El síndrome de Usher es una enfermedad genética poco común que combina pérdida auditiva con alteraciones visuales progresivas. Su rasgo más característico en el ámbito oftalmológico es la aparición de una retinopatía hereditaria, conocida como retinitis pigmentosa, que deteriora de manera lenta pero constante las células fotorreceptoras de la retina. Estas células son las encargadas de transformar la luz en señales eléctricas que el cerebro interpreta como imágenes, por lo que su daño afecta directamente la capacidad de ver.
En las primeras etapas, las personas con síndrome de Usher suelen notar dificultades para ver en lugares con poca luz (nictalopía) o problemas para adaptarse a la oscuridad después de estar expuestos a la claridad. Con el tiempo, el campo visual se va reduciendo, dando lugar a lo que se conoce como visión en túnel, una sensación de ver como a través de un tubo o un pequeño círculo central.
El ritmo de progresión puede variar según el tipo de síndrome y el gen afectado, pero en la mayoría de los casos la pérdida visual tiende a avanzar con los años. Este proceso puede influir en la orientación, la movilidad y la independencia del paciente, especialmente en etapas adultas.
Aunque el síndrome de Usher también compromete la audición, la salud visual ocupa un lugar central en su manejo, ya que la detección precoz de los signos retinianos permite ofrecer tratamientos y ayudas visuales que mejoran la calidad de vida. La participación de un oftalmólogo especializado en retina es esencial para monitorear la evolución y orientar al paciente sobre las mejores opciones de cuidado ocular.
Síndrome de Usher y genética
El síndrome de Usher tiene un origen genético claramente identificado. Se transmite de forma autosómica recesiva, lo que significa que ambos padres son portadores de una copia alterada del gen, aunque no presenten síntomas. Cuando el hijo hereda las dos copias defectuosas, se desarrolla la enfermedad.
Desde el punto de vista visual, estas mutaciones genéticas provocan anomalías en las proteínas esenciales para el funcionamiento de las células fotorreceptoras de la retina (los bastones y conos). Estas células son responsables de captar la luz y el color, y enviarlas al cerebro a través del nervio óptico. Cuando las proteínas no funcionan correctamente, las células comienzan a degenerarse, lo que explica la pérdida progresiva de visión que caracteriza al síndrome.
Entre los genes más estudiados se encuentran MYO7A, USH2A, CLRN1, CDH23 y PCDH15, cada uno relacionado con diferentes tipos de Usher. Las mutaciones en estos genes alteran la estructura y estabilidad de las células sensoriales del oído interno y de la retina, generando una afectación simultánea en ambos sistemas.
En la actualidad, los avances en genética permiten no solo confirmar el diagnóstico, sino también determinar el tipo específico de síndrome de Usher que presenta el paciente. Este conocimiento es clave porque la identificación del gen afectado puede orientar el pronóstico visual y abrir la puerta a futuras terapias génicas destinadas a frenar la degeneración retiniana.
Si bien todavía no existe una cura definitiva, el estudio genético se ha convertido en una herramienta indispensable dentro del cuidado oftalmológico, ya que permite planificar un seguimiento más preciso, anticipar complicaciones visuales y personalizar las estrategias de tratamiento para cada paciente.
Tipos de síndrome de Usher
Aunque todos los casos de síndrome de Usher comparten el origen genético y la combinación de pérdida auditiva con alteraciones visuales, no todos se manifiestan de la misma forma. Existen tres tipos principales —I, II y III— que se diferencian por la edad de aparición, la severidad de los síntomas y la velocidad con la que progresa la pérdida de visión.
Tipo I
Es la forma más severa del síndrome. Los niños con este tipo suelen nacer con sordera profunda y presentan dificultades en el equilibrio debido a alteraciones en el oído interno. A nivel ocular, la retinitis pigmentosa se manifiesta desde etapas tempranas de la infancia, ocasionando una rápida pérdida de la visión periférica. En muchos casos, los primeros signos aparecen antes de los 10 años, con dificultades para ver en ambientes oscuros o adaptarse a cambios de iluminación.
Con el tiempo, la degeneración retiniana puede avanzar hacia el centro de la visión, limitando de manera significativa la capacidad para realizar actividades cotidianas como leer o reconocer rostros.
Tipo II
Este tipo es más frecuente y se caracteriza por una pérdida auditiva moderada a severa desde el nacimiento, pero sin afectación del equilibrio. En el ámbito visual, los síntomas suelen aparecer durante la adolescencia o adultez temprana, cuando el paciente empieza a notar problemas de visión nocturna o una reducción del campo visual.
La progresión de la retinitis pigmentosa en este grupo suele ser más lenta, pero igualmente requiere controles oftalmológicos periódicos para monitorear el avance de la degeneración retiniana.
Tipo III
Menos común que los anteriores, el tipo III se caracteriza por una pérdida auditiva y visual progresiva, que puede iniciar en la infancia, adolescencia o incluso en la adultez. En estos casos, la retina puede mantener una función aceptable por varios años, pero los síntomas visuales se van acentuando con el paso del tiempo.
Debido a su evolución variable, los pacientes con tipo III necesitan un seguimiento oftalmológico personalizado, ya que la rapidez de la degeneración visual puede diferir notablemente de una persona a otra.
En todos los tipos, la afectación de la retina representa el principal desafío visual. La detección temprana de los cambios retinianos mediante pruebas especializadas, como el electrorretinograma (ERG) o la tomografía de coherencia óptica (OCT), permite establecer un plan de manejo adecuado para conservar la mayor agudeza visual posible.
▶ ¿Necesitas una evaluación especializada? Reserva una cita en Opeluce.
Causas del síndrome de Usher
El síndrome de Usher es una enfermedad de origen genético causada por mutaciones en ciertos genes que intervienen en el funcionamiento del oído interno y la retina. Estas alteraciones impiden que las células sensoriales cumplan correctamente su función, provocando tanto pérdida auditiva como visual.
Las principales causas relacionadas son:
- Mutaciones genéticas hereditarias transmitidas por ambos padres portadores, aunque ellos no presenten síntomas.
- Degeneración progresiva de las células fotorreceptoras de la retina, responsables de captar la luz y las imágenes.
- Alteraciones en proteínas esenciales para mantener la estructura y función de la retina.
- Factores ambientales secundarios, como la exposición solar excesiva sin protección o el tabaquismo, que pueden acelerar el daño visual.
Si bien el componente genético es la base del síndrome, la evolución ocular varía entre pacientes. Por ello, los controles oftalmológicos periódicos son esenciales para evaluar la progresión de la enfermedad y proteger la salud visual.
▶ Agenda tu cita con nosotros y protege tu salud ocular hoy
Síntomas del síndrome de Usher
Los síntomas del síndrome de Usher pueden variar según el tipo y la severidad de la enfermedad, pero en todos los casos existe una afectación progresiva de la visión que suele comenzar en etapas tempranas de la vida. En la mayoría de los pacientes, las manifestaciones visuales son consecuencia directa de la retinitis pigmentosa, una alteración degenerativa de la retina que deteriora gradualmente las células encargadas de captar la luz.
Los principales signos oculares incluyen:
- Dificultad para ver en lugares con poca luz o de noche (nictalopía). Este suele ser uno de los primeros síntomas visuales.
- Pérdida progresiva del campo visual periférico, conocida como visión en túnel, que genera la sensación de ver a través de un espacio reducido.
- Problemas de adaptación a los cambios de iluminación, por ejemplo, al pasar de un ambiente oscuro a uno iluminado o viceversa.
- Disminución de la agudeza visual con el paso del tiempo, especialmente en las fases más avanzadas.
- En algunos casos, sensibilidad a la luz intensa (fotofobia) o dificultad para distinguir colores.
La progresión visual puede ser lenta y pasar inadvertida durante varios años, lo que retrasa el diagnóstico. Por ello, cualquier señal de disminución de la visión nocturna o pérdida de visión lateral debe ser motivo para acudir a una evaluación oftalmológica especializada.
A medida que la enfermedad avanza, los pacientes pueden presentar limitaciones para orientarse, leer o reconocer rostros, lo que afecta su independencia y calidad de vida. Sin embargo, un diagnóstico temprano y un seguimiento regular permiten implementar medidas que ayudan a conservar la función visual durante más tiempo.
Diagnóstico del síndrome de Usher
El diagnóstico del síndrome de Usher se basa en la combinación de evaluaciones auditivas y, sobre todo, oftalmológicas, ya que los cambios en la retina suelen ser determinantes para confirmar la enfermedad.
En la revisión ocular, el fondo de ojo permite detectar signos característicos de retinitis pigmentosa, como pigmentos oscuros y adelgazamiento de los vasos retinianos. Además, pruebas como la tomografía de coherencia óptica (OCT) y el electrorretinograma (ERG) permiten evaluar el grado de daño en las células fotorreceptoras y monitorear la evolución visual con precisión.
Las pruebas genéticas complementan el diagnóstico al identificar el gen alterado responsable de la enfermedad, ayudando a orientar el pronóstico y brindar asesoramiento familiar.
Realizar evaluaciones oftalmológicas periódicas es esencial para detectar el síndrome de Usher a tiempo y actuar antes de que la pérdida visual avance. Contar con el apoyo de un centro especializado como Opeluce, que dispone de tecnología avanzada y especialistas en retina, garantiza un diagnóstico confiable y un seguimiento visual integral.
Tratamiento del síndrome de Usher
Aunque el síndrome de Usher no tiene una cura definitiva, los avances médicos han permitido desarrollar estrategias que ayudan a ralentizar la progresión visual y mejorar la calidad de vida de los pacientes. El objetivo principal del tratamiento es preservar la función de la retina y mantener la mayor capacidad visual posible durante el mayor tiempo.
Entre las medidas más importantes se incluyen
- Suplementos y protección ocular: en algunos casos, el uso controlado de antioxidantes (como la vitamina A, bajo indicación médica) puede contribuir a proteger las células fotorreceptoras. Además, se recomienda el uso diario de gafas con filtro UV para evitar daños adicionales por la radiación solar.
- Ayudas ópticas y rehabilitación visual: existen dispositivos y técnicas que facilitan la lectura, la orientación y las tareas cotidianas, como lupas electrónicas, filtros especiales y programas de entrenamiento visual.
- Terapias génicas e investigaciones: actualmente se desarrollan tratamientos experimentales que buscan reemplazar o corregir los genes defectuosos responsables de la degeneración retiniana. Aunque aún se encuentran en fase de estudio, representan una esperanza para el futuro del manejo visual en pacientes con síndrome de Usher.
- Control oftalmológico regular: los exámenes periódicos permiten detectar de manera temprana cualquier cambio en la retina y adaptar las estrategias de manejo a la evolución del paciente.
El acompañamiento por parte de un oftalmólogo especializado en retina es fundamental para ajustar el tratamiento de forma individualizada y orientar al paciente sobre los cuidados más adecuados.
Preguntas frecuentes sobre el síndrome de Usher
¿A qué edad aparecen los primeros síntomas visuales?
Depende del tipo de síndrome de Usher. En los casos más severos, los síntomas oculares pueden comenzar en la niñez, mientras que en otros tipos aparecen durante la adolescencia o la adultez temprana. En general, los primeros signos suelen ser dificultad para ver en lugares oscuros y una visión lateral reducida.
¿El síndrome de Usher puede causar ceguera total?
La pérdida de visión varía entre pacientes. En la mayoría de los casos, la degeneración de la retina provoca una visión en túnel o una disminución importante del campo visual, pero muchas personas conservan visión útil por varios años, especialmente si reciben seguimiento y cuidados oftalmológicos adecuados.
¿Por qué es importante realizar evaluaciones oftalmológicas periódicas?
Porque los cambios en la retina pueden detectarse incluso antes de que el paciente note los síntomas. Los controles visuales regulares permiten actuar a tiempo, proteger las áreas retinianas funcionales y adaptar las estrategias de tratamiento según la evolución.
¿Dónde puedo acudir para una evaluación especializada?
En Opeluce, contamos con tecnología avanzada y especialistas en retina capaces de detectar enfermedades hereditarias como el síndrome de Usher en sus etapas iniciales. Un diagnóstico temprano marca la diferencia en la preservación de la visión y en la planificación de un tratamiento integral.
La importancia de cuidar la visión ante el síndrome de Usher
El síndrome de Usher representa un desafío tanto para la audición como para la vista, pero es en la salud ocular donde la detección temprana puede marcar una diferencia decisiva. Identificar los primeros signos de pérdida visual y mantener un control constante con un especialista en retina son pasos fundamentales para preservar la visión y la autonomía del paciente.
Aunque actualmente no exista una cura definitiva, los avances en diagnóstico, rehabilitación y genética ofrecen cada vez más herramientas para mejorar la calidad de vida. El acompañamiento médico, la educación visual y la orientación profesional permiten que las personas con esta condición se adapten mejor a los cambios y mantengan una visión funcional durante más tiempo.
Si notas dificultad para ver de noche, reducción del campo visual o tienes antecedentes familiares de enfermedades oculares hereditarias, no esperes más. Agenda una evaluación especializada en Opeluce y recibe atención personalizada de expertos en retina y diagnóstico ocular avanzado. Cuidar tu visión hoy puede hacer una gran diferencia mañana.
📞 Reserva tu cita llamando al (01) 206-4700
📍 Estamos ubicados en Av. Arequipa 1885 Lince – Lima